
Why Geometry Matters in Graph Neural Networks
Published:
The Geometric Setting
Intuition First: Imagine you have a molecular model kit. You can describe the connectivity — carbon bonded to two oxygens — without saying how those bonds are arranged in 3D space. But a flat (180°) CO₂ and a bent (120°) arrangement have wildly different properties. Standard GNNs only read the assembly instructions; geometric GNNs also read the 3D blueprint.
Consider a molecule modelled as a graph G = (V, E, X, R):
- V: atoms (nodes), E: bonds (edges), X: atomic features (atom type, charge)
- R ∈ ℝ^{N×3}: 3D coordinates of each atom
Standard GNNs use only (V, E, X) and ignore R. This loses crucial information:
Stereoisomers: molecules with the same atoms and bonds but different 3D arrangement. L-alanine and D-alanine are mirror images — identical connectivity, completely different biological activity. A GNN without 3D coordinates assigns them the same embedding.
Conformation: proteins fold into specific 3D shapes that determine their function. Two proteins with the same sequence but different folds (conformers) have different biological roles — invisible to connectivity-only GNNs.
Distances and angles: in chemistry, reaction rates depend on bond angles and dihedral angles — geometric properties that cannot be inferred from connectivity alone.
The Symmetry Problem
3D coordinates are not unique to a molecule:
- Translation: moving the molecule in space leaves chemistry unchanged
- Rotation: rotating the molecule leaves chemistry unchanged
- Reflection: mirroring leaves chemistry unchanged for most properties (but not chirality)
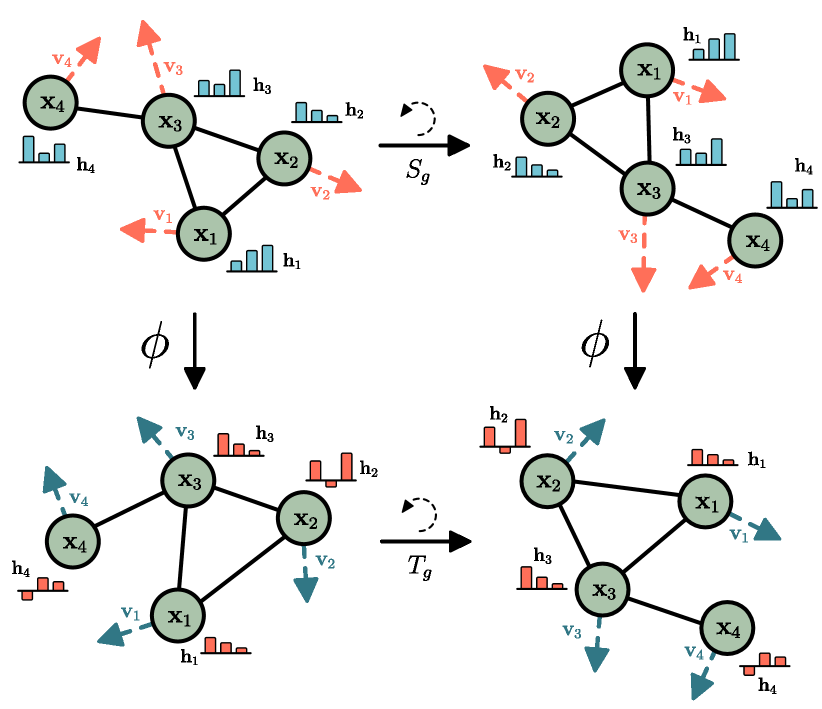
A model that takes 3D coordinates as input must respect these symmetries — its output should not change when we translate, rotate, or (for most properties) reflect the molecule.
Failure mode: naive addition of coordinates to node features gives the model different inputs for the same molecule in different orientations. The model must learn the symmetry from data — requiring enormous amounts of training examples covering all orientations.
Invariance vs Equivariance
Invariant: f(T(G)) = f(G) for all symmetry transformations T. The output is unchanged.
For graph-level properties (energy, solubility): the property is invariant. Rotating the molecule doesn’t change its energy.
Equivariant: f(T(G)) = T(f(G)). The output transforms the same way as the input.
For node-level vector properties (forces, velocities): the property is equivariant. If we rotate the molecule, the forces rotate the same way.
What Standard GNNs Cannot Do
| Task | Requires | Standard GNN |
|---|---|---|
| Distinguish stereoisomers | 3D chirality | Cannot |
| Predict 3D forces | Equivariant vectors | Cannot |
| Learn protein structure | 3D coordinates + symmetry | Cannot |
| Model crystal symmetry | Space group symmetry | Cannot |
| Point cloud processing | 3D position | Cannot |
What Geometric GNNs Add
Three levels of geometric sophistication:
Level 1: Distance-based (invariant) Add interatomic distances ||r_u - r_v|| as edge features. The model is invariant to translation and rotation (distances are invariant) but cannot predict vector quantities.
Level 2: Angle-based (richer invariant) Add angles between bond triplets (u-v-w) and dihedral angles (u-v-w-x). DimeNet, SphereNet operate at this level.
Level 3: Equivariant (full 3D) Process 3D vectors as vectors — not just their magnitudes. EGNN, SE(3)-Transformers, NequIP, MACE operate at this level.
Real Applications
Drug discovery: predict binding affinity, toxicity, ADMET properties from 3D molecular structure.
Protein structure prediction: model protein folding and protein-protein interaction geometry.
Materials science: predict crystal properties (band gap, stability) from atomic positions in unit cell.
Robotics: process point cloud sensor data while maintaining rotational equivariance.
Particle physics: predict particle interaction properties with detector geometry.
Summary
Adding geometry to GNNs is not optional for applications where 3D structure matters. The challenge is doing so while respecting the symmetries of 3D space — translation, rotation, reflection. The subsequent posts in this section cover the architectures (EGNN, SE(3)-Transformers, TFN) that solve this systematically using group theory.
References
- Bronstein, M. M., Bruna, J., Cohen, T., & Veličković, P. (2021). Geometric Deep Learning: Grids, Groups, Graphs, Geodesics, and Gauges. arXiv 2021 (the unifying geometric deep learning blueprint: symmetry groups, equivariance, and the 5G framework).
- Schütt, K. T., Kindermans, P.-J., Sauceda Felix, H. E., Chmiela, S., Tkatchenko, A., & Müller, K.-R. (2017). SchNet: A Continuous-Filter Convolutional Neural Network for Modeling Quantum Interactions. NeurIPS 2017 (SchNet: distance-based interaction filters for molecular property prediction).
- Klicpera, J., Groß, J., & Günnemann, S. (2020). Directional Message Passing for Molecular Graphs. ICLR 2020 (DimeNet: bond angles enable chirality-aware message passing beyond pure distances).