
GNNs for Molecules: Drug Discovery and Material Design
Published:
The Drug Discovery Pipeline
Intuition First: Finding a drug is like searching for a key that fits a specific lock (the protein target). Chemical space contains roughly 10^60 possible drug-like molecules — far too many to test physically. A GNN is trained on known key–lock pairs to predict which untested keys are likely to fit. It learns that certain atom arrangements near certain bond types correlate with good binding — then uses those patterns to score billions of virtual molecules in seconds rather than years.
Drug discovery takes 10-15 years and $2B+ per approved drug. GNNs accelerate three key stages:
- Virtual screening: filter billions of candidate molecules to thousands using property predictions
- Lead optimisation: predict ADMET (absorption, distribution, metabolism, excretion, toxicity) properties
- De novo design: generate novel molecules with desired properties
What GNNs Predict
ADMET properties:
- Solubility: how much dissolves in water (affects bioavailability)
- Lipophilicity (LogP): determines membrane permeability
- Toxicity (hERG, AMES): cardiac toxicity, mutagenicity
- Metabolic stability: how quickly the liver degrades the drug
- Blood-brain barrier penetration: reaches the brain?
Binding affinity:
- Predicted IC50, Ki, Kd for specific protein targets
- Virtual screening: rank candidates by predicted affinity
Quantum chemistry (QM9 benchmarks):
- HOMO-LUMO gap (electronic excitation energy)
- Dipole moment, polarisability
- Zero-point energy
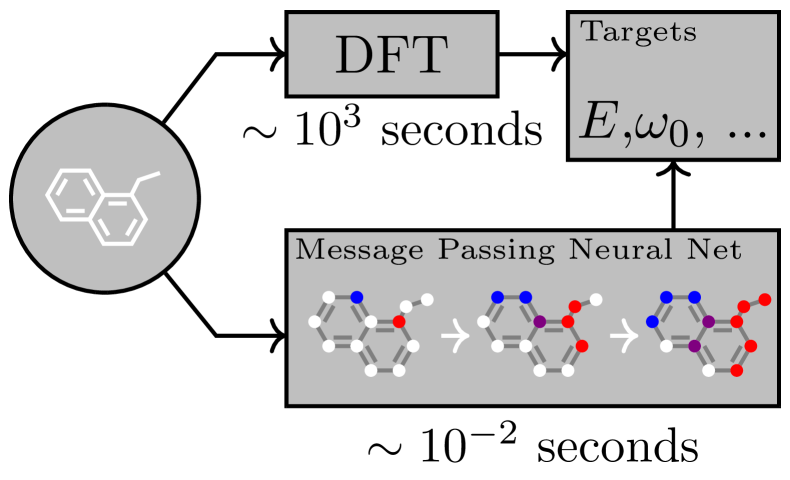
The GNN Pipeline for Molecules
SMILES string → RDKit graph → Atom/bond features
↓
GNN (2-4 layers)
↓
Node embeddings
↓
Global pooling (sum/attention)
↓
Graph embedding
↓
MLP → property prediction
Atom features: atomic number, formal charge, number of Hs, hybridisation (sp/sp²/sp³), aromaticity, chirality
Bond features: bond type (single/double/triple/aromatic), is-conjugated, is-ring, stereo
Key Models for Molecular Property Prediction
MPNN (Gilmer et al., 2017): introduced the message passing neural network framework for molecules. First systematic study showing GNNs outperform Morgan fingerprints on QM9.
AttentiveFP (Xiong et al., 2019): adds graph attention for molecular property prediction. Handles multi-task learning across different ADMET endpoints.
Grover (Rong et al., 2020): self-supervised pre-training on 10M unlabelled molecules, then fine-tune on small labelled datasets. Solves the labelled data scarcity problem in drug discovery.
MolBERT / ChemBERTa: treat SMILES as a sequence, apply BERT-style pre-training. Competitive with graph-based methods on many benchmarks.
Virtual Screening at Scale
The challenge: DrugBank has 13,000 approved drugs. PubChem has 100M compounds. Synthesisable chemical space has ~10^{60} molecules. Which to test?
GNN-based screening:
- Train GNN on known active/inactive pairs for target protein
- Run inference on virtual library (billions of molecules)
- Select top-k predicted actives for experimental validation
Companies like Insilico Medicine, Schrödinger, and Recursion use GNN-based virtual screening as a core workflow.
Protein-Ligand Interaction
Beyond single-molecule property prediction: predicting how a small molecule (ligand) binds to a protein target.
Input: protein structure (graph of residues) + ligand structure (graph of atoms) + 3D binding pose
Model: heterogeneous GNN with protein nodes, ligand nodes, and protein-ligand interaction edges. Equivariant GNNs (EGNN, SE3-Transformers) respect 3D symmetry.
Output: binding affinity score (docking score)
Benchmarks
- MoleculeNet: 17 datasets covering classification and regression across ADMET endpoints
- OGB-molhiv: HIV activity (41,127 molecules)
- OGB-molpcba: 128 PCBA assays (437,929 molecules)
- QM9: 12 quantum chemistry properties (134k molecules)
- MD17: molecular dynamics trajectories for force field learning
Summary
GNNs have become the default molecular representation learning method in computational drug discovery, replacing handcrafted Morgan fingerprints. The key advantages: end-to-end learning, generalisation across chemical space, and compatibility with both 2D connectivity and 3D geometric information. With pre-training on large unlabelled databases, GNN-based models now approach expert-level performance on standard ADMET prediction benchmarks.
References
- Gilmer, J., Schütt, K. T., Ramsundar, B., Ramakrishnan, R., Bronskill, M., Gomes, C., & Dahl, G. E. (2017). Neural Message Passing for Quantum Chemistry. ICML 2017 (MPNN: unified framework for molecular GNNs, benchmarked on QM9 properties).
- Rong, Y., Bian, Y., Xu, T., Xie, W., Wei, Y., Huang, W., & Huang, J. (2020). Self-Supervised Graph Transformer on Large-Scale Molecular Data. NeurIPS 2020 (GROVER: large-scale pre-training of molecular GNNs on 10M unlabelled molecules for drug property prediction).
- Hu, W., Liu, B., Gomes, J., Zitnik, M., Liang, P., Pande, V., & Leskovec, J. (2020). Strategies for Pre-training Graph Neural Networks. ICLR 2020 (systematic study of GNN pre-training strategies for molecular property prediction and other biological tasks).