
Molecular GNNs: Learning on Atoms and Bonds
Published:
Molecules as Graphs
A molecule G = (V, E, X, R) where:
- V: atoms (carbon, oxygen, nitrogen, …)
- E: bonds (single, double, triple, aromatic)
- X: atom features (atomic number, charge, hybridisation, …)
- R ∈ ℝ^{N×3}: 3D coordinates (from DFT calculations or conformation search)
The task: predict molecular properties from G. Properties include:
- HOMO-LUMO gap (electronic structure, relevant to photovoltaics)
- Solubility (pharmaceutical drug delivery)
- Toxicity (drug safety screening)
- Binding affinity (protein-drug interaction)
- Dipole moment, polarisability (material properties)
From Fingerprints to GNNs
Intuition First: Morgan fingerprints work like a census of neighbourhoods. Each atom looks at the atoms within K bonds of it, hashes the whole pattern to a number, and reports that number. The GNN approach instead lets atoms talk to their neighbours iteratively — at round 1 atoms share their own identity, at round 2 they share what they heard from round 1, and so on. Unlike the fixed hash, GNN representations are learned end-to-end for the specific task, so they focus on the features that actually predict toxicity (or solubility, or binding), rather than encoding everything uniformly.
Traditional approach — Morgan fingerprints (ECFP):
- Encode each atom’s K-hop neighbourhood as a hash
- Sum over all atoms → fixed-size bit vector
- Feed to SVM or random forest
GNN approach:
- Run K rounds of message passing → node embeddings encode K-hop neighbourhoods
- Global pooling → graph embedding
- MLP → property prediction
GNNs outperform fingerprints because they learn task-specific features rather than encoding all structural information uniformly.
Level 1: 2D GNNs (Connectivity Only)
Standard GCN, GAT, or GIN on the molecular graph with:
- Node features: atomic number (one-hot), formal charge, number of Hs, hybridisation
- Edge features: bond type (single/double/triple/aromatic), is-conjugated, is-ring
Examples: MPNN (Gilmer et al., 2017), AttentiveFP.
Limitation: cannot distinguish stereoisomers (L-alanine vs D-alanine have identical connectivity). Missing 3D information.
Level 2: 3D Distance-Based (Invariant)
Add interatomic distances as edge features. Build a graph where all atoms within cutoff distance r_c are connected (not just bonded atoms).
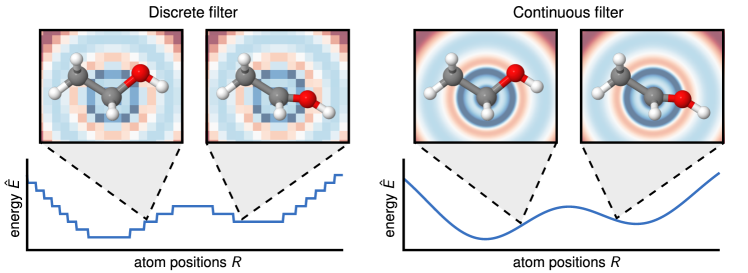
SchNet (Schütt et al., 2017): uses continuous-filter convolutions based on radial basis functions of distance:
Where W is a distance-dependent filter network. This achieves rotational invariance (distances are invariant) but cannot detect angles.
QM9 performance: SchNet achieves chemical accuracy on several QM9 targets (energy, HOMO energy, LUMO energy) — competitive with DFT at orders-of-magnitude less compute.
Level 3: Angular GNNs (Bond Angles)
Adding interatomic distances is not sufficient — two conformers can have identical pairwise distance matrices but different angles. DimeNet incorporates angles between bond triplets.
DimeNet (Klicpera et al., 2020): messages are defined over directed edges (not just nodes), including the angle between edges:
Where θ_{kji} is the angle at j between bonds ji and jk. DimeNet uses Bessel functions for radial features and spherical harmonics for angular features.
SphereNet: adds dihedral angles (torsions) — the angle between two planes defined by four atoms. This completes the geometric description of local 3D structure.
Level 4: Equivariant GNNs
Equivariant models process 3D positions as vectors, maintaining E(n) or SE(3) equivariance. They can predict both scalar properties (energy) and vector properties (forces) without violating symmetry.
EGNN: distance-based messages + equivariant coordinate updates. Simple, fast, effective for energy prediction.
NequIP: TFN-style tensor features + message passing. State-of-the-art for force fields with few training points.
MACE: many-body interactions. Current SOTA on MD17 molecular dynamics benchmark.
Benchmarks
QM9: 134k small organic molecules (up to 9 heavy atoms). 12 quantum chemical properties (HOMO energy, LUMO energy, dipole moment, etc.) computed by DFT.
MD17: molecular dynamics trajectories. Predict energy and forces at each timestep. Tests generalisation to conformational space.
OGB-molhiv / OGB-molpcba: large-scale drug discovery benchmarks (41k/437k molecules).
PDBbind: protein-ligand binding affinity from crystal structures.
The Accuracy Progression on QM9 (HOMO-LUMO gap)
Morgan fingerprint + RF: ~0.5 eV
MPNN (2D): ~0.18 eV
SchNet (distance): ~0.07 eV
DimeNet (angles): ~0.05 eV
SphereNet (dihedrals): ~0.03 eV
NequIP (equivariant): ~0.02 eV
MACE (many-body): ~0.01 eV (near DFT accuracy)
Each geometric level roughly halves the error. Chemical accuracy is ~0.04 eV — equivariant models are within or below this threshold.
Summary
| Level | Geometry used | Key model | QM9 error | ||||
|---|---|---|---|---|---|---|---|
| 2D (connectivity) | None | MPNN, GIN | ~0.18 eV | ||||
| Distances | r_ij | SchNet | ~0.07 eV | ||||
| Distances + angles | θ_{ijk} | DimeNet | ~0.05 eV | ||||
| Distances + angles + dihedrals | φ_{ijkl} | SphereNet | ~0.03 eV | ||||
| Full equivariance | 3D vectors | NequIP, MACE | ~0.01 eV |
For industrial drug discovery, 2D GNNs suffice for fast virtual screening. For physics-accurate property prediction and force fields, equivariant models are the only option.
References
- Gilmer, J., Schütt, K. T., Ramsundar, B., Ramakrishnan, R., Bronskill, M., Gomes, C., & Dahl, G. E. (2017). Neural Message Passing for Quantum Chemistry. ICML 2017 (MPNN: unified message passing framework for quantum chemistry, benchmarked on QM9).
- Schütt, K. T., Kindermans, P.-J., Sauceda Felix, H. E., Chmiela, S., Tkatchenko, A., & Müller, K.-R. (2017). SchNet: A Continuous-Filter Convolutional Neural Network for Modeling Quantum Interactions. NeurIPS 2017 (SchNet: continuous-filter convolutions over interatomic distances for E(3)-invariant molecular property prediction).
- Klicpera, J., Groß, J., & Günnemann, S. (2020). Directional Message Passing for Molecular Graphs. ICLR 2020 (DimeNet: adds bond angles to message passing, enabling chirality-aware representations beyond pure distances).
- Liu, Y., Wang, L., Liu, M., Lin, Y., Zhang, X., Oztekin, B., & Ji, S. (2022). Spherical Message Passing for 3D Molecular Graphs. ICLR 2022 (SphereNet: extends DimeNet with torsion angles for full 3D geometry encoding).